


CRA(臨床開発モニター)は、治験を実施する際に遵守すべき基準であるGCPや治験の計画書に従って、治験が適切に行われるように監視(モニタリング)する仕事です。製薬会社や病院で働くさまざまな職種の方と協力して業務を行います。
この記事では、現役のCRA(臨床開発モニター)の声や現場の裏話、写真やイラストを交えて、CRA(臨床開発モニター)の仕事内容を分かりやすく解説しています。「CRA(臨床開発モニター)って何をする人?」「CRA(臨床開発モニター)の仕事内容が分からない!」という方は、ぜひご覧ください。
※CRAの仕事内容とやりがいについて、音声で分かりやすく解説します。
一目で分かる! 構造化された要約を見る
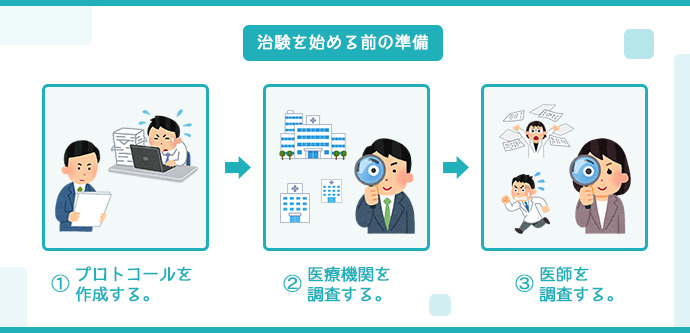
 治験開始前の準備
治験開始前の準備









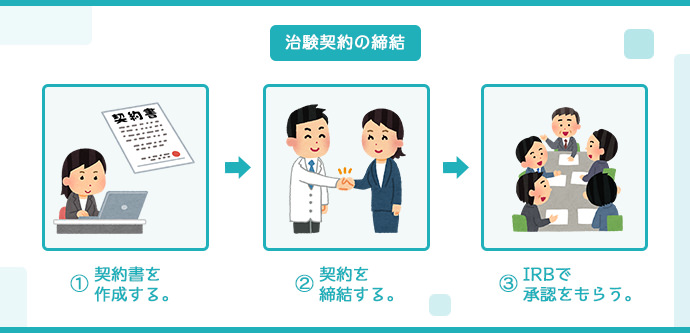
 治験契約の締結
治験契約の締結





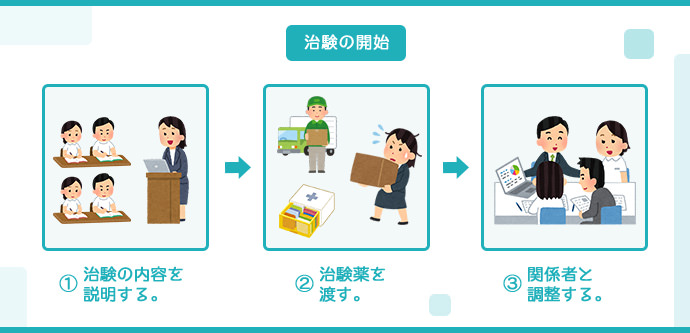
 治験の開始
治験の開始


 各関係者と打ち合わせが必要な内容
各関係者と打ち合わせが必要な内容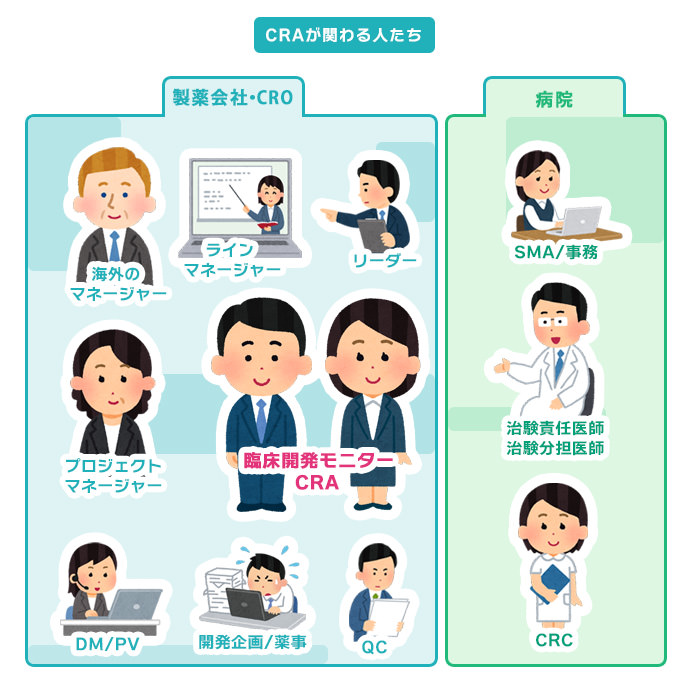
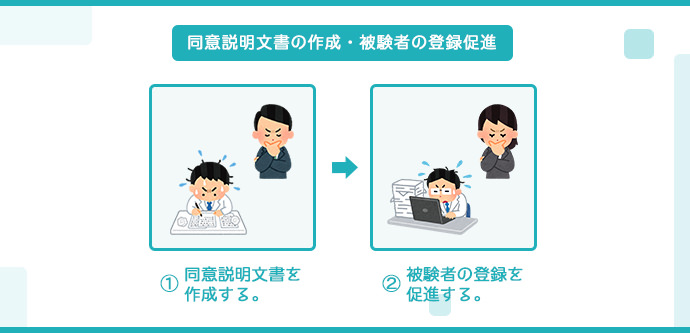
 同意説明文書の作成・被験者の登録促進
同意説明文書の作成・被験者の登録促進


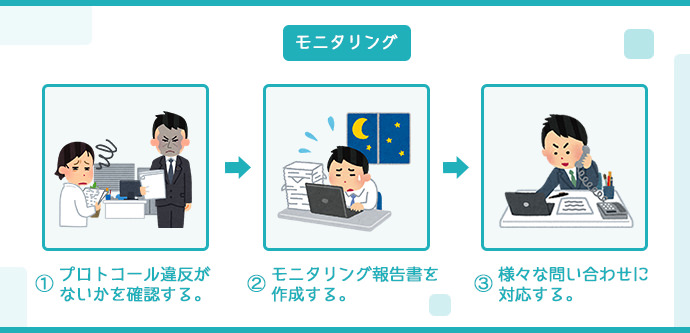
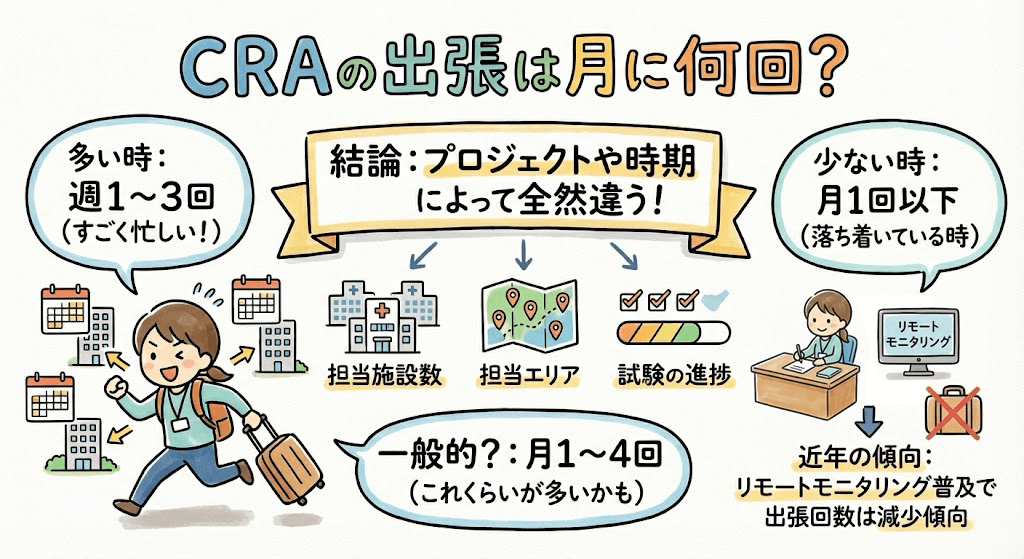
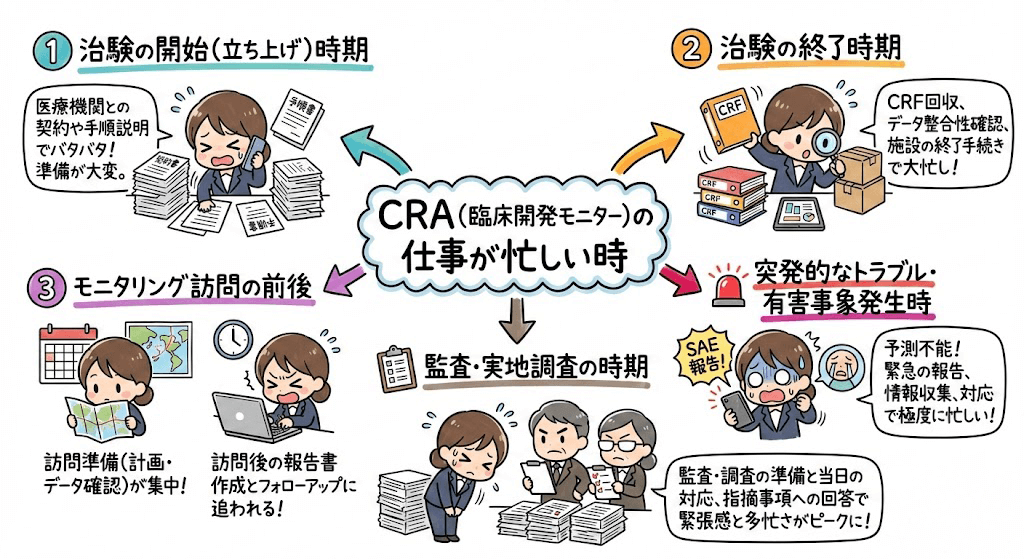
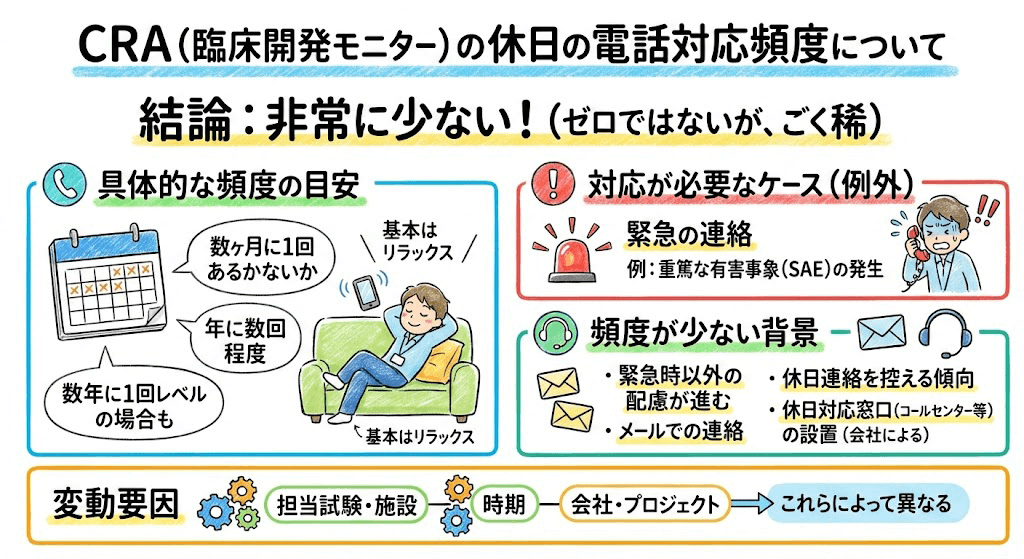
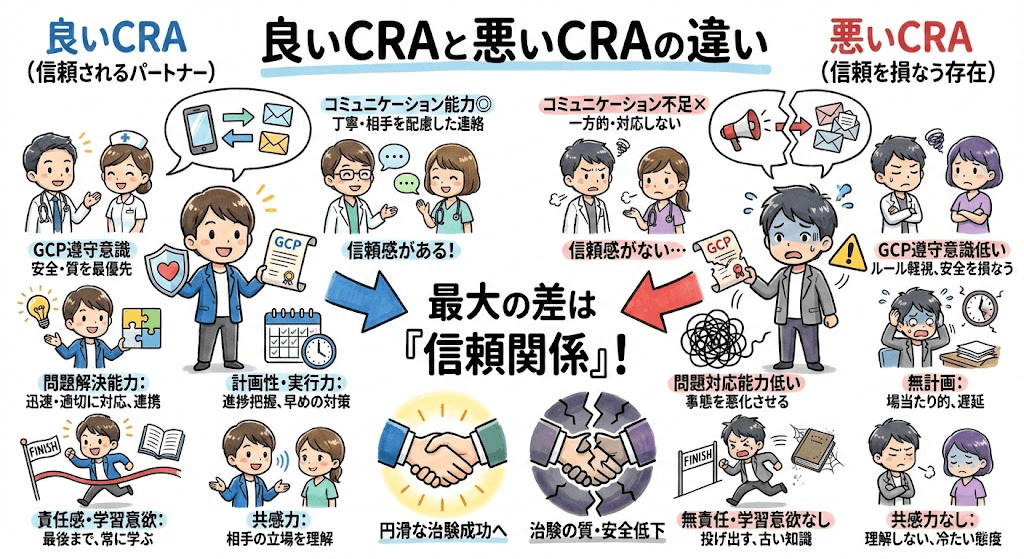
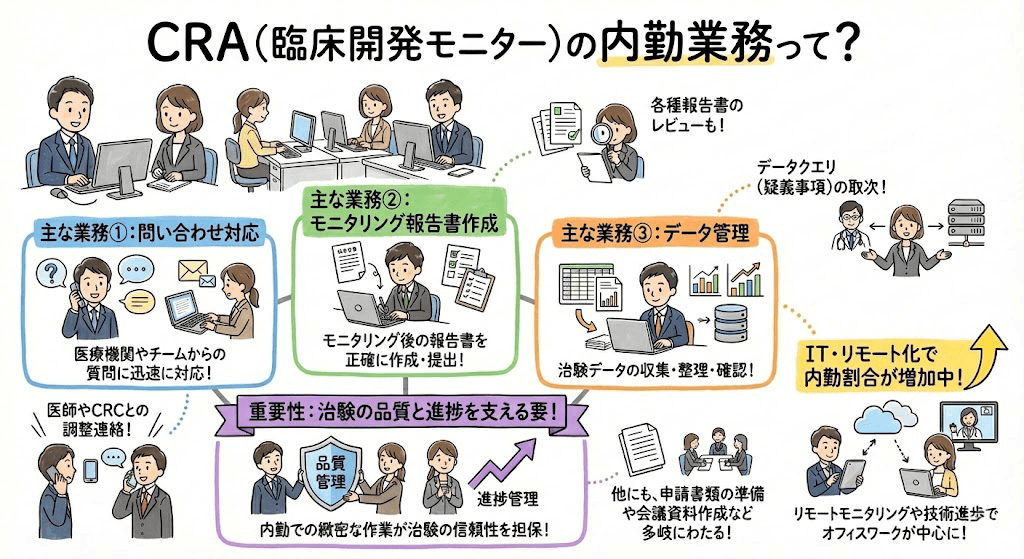
 モニタリング
モニタリング

 プロトコール(治験実施計画書)違反の主な調査項目
プロトコール(治験実施計画書)違反の主な調査項目




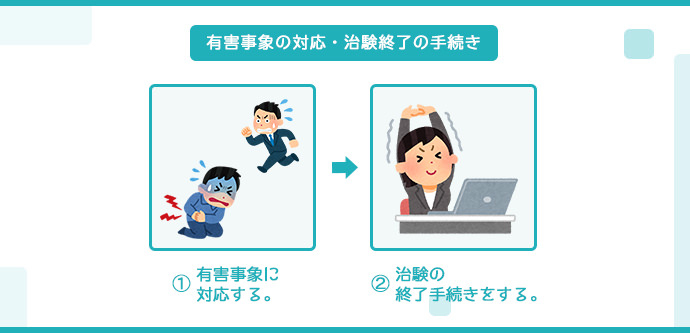
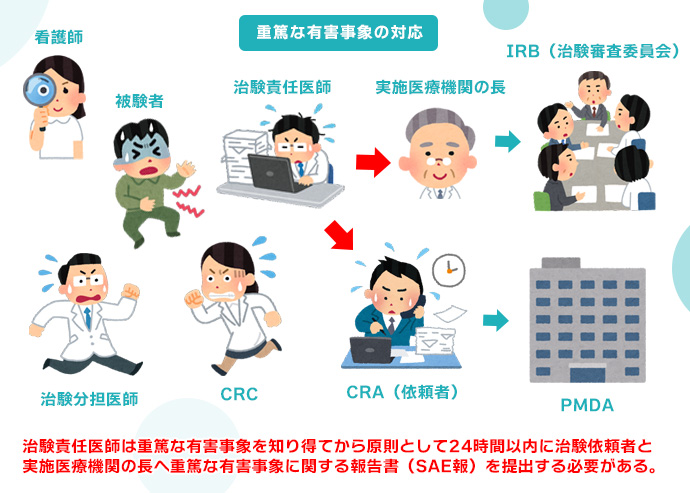
 有害事象と文書改訂の対応・治験の終了
有害事象と文書改訂の対応・治験の終了




 よくある質問とみんなの回答
よくある質問とみんなの回答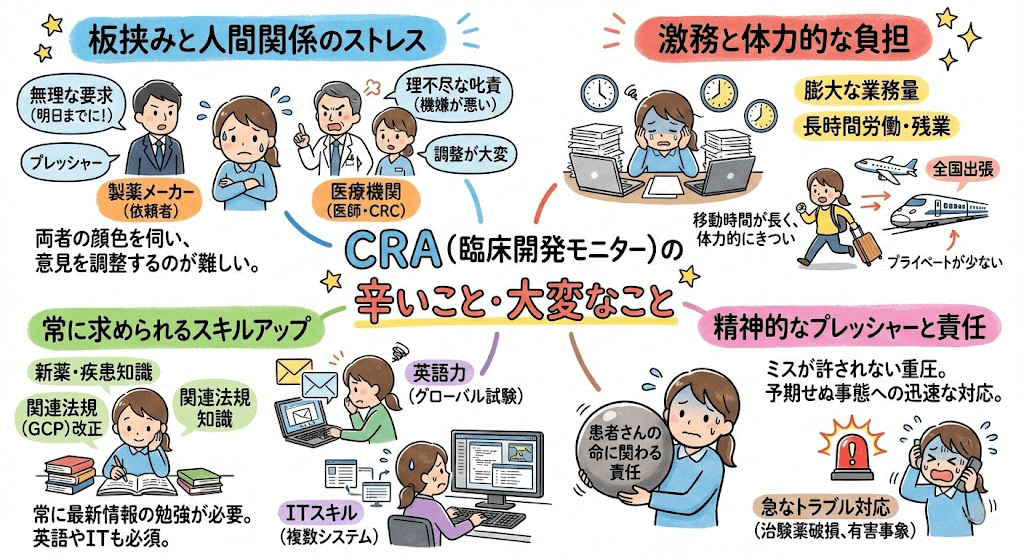












 年収査定はこちら
年収査定はこちら
 合格予想はこちら
合格予想はこちら
 掲示板で質問をする
掲示板で質問をする







 CRA
CRA

 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAに
CRAに
 CRAの
CRAの
 CRO
CRO
 CRO
CRO
 臨床開発
臨床開発
 製薬会社と
製薬会社と
 CROから
CROから



 2027年4月からの転職
2027年4月からの転職 CRA未経験特集
CRA未経験特集 薬剤師特集
薬剤師特集 MR特集
MR特集 看護師特集
看護師特集 臨床検査技師特集
臨床検査技師特集 保健師特集
保健師特集 獣医師特集
獣医師特集 理系大卒・院卒特集
理系大卒・院卒特集 CRC経験者特集
CRC経験者特集


 求人検索
求人検索  ログイン
ログイン 会員さま専用
会員さま専用 CRAの仕事
CRAの仕事  臨床開発業界の研究
臨床開発業界の研究 経験・資格別の注意点
経験・資格別の注意点 応募書類の作成
応募書類の作成 面接・適性検査の対策
面接・適性検査の対策 みんなのクチコミ
みんなのクチコミ みんなの質問と回答
みんなの質問と回答 転職成功事例
転職成功事例 マンガで分かるCRA
マンガで分かるCRA 便利な機能
便利な機能 相談/年収査定/合格予想
相談/年収査定/合格予想 2027年から働くには?
2027年から働くには? 退職手続き
退職手続き 開催中のキャンペーン
開催中のキャンペーン 《CRAばんく》とは
《CRAばんく》とは