

CRA(臨床開発モニター)の「臨床開発」とは、どのような意味なのか正しく理解していますか? CRA(臨床開発モニター)になるためには、臨床開発とは何かを正しく理解しておくことが必要です。
臨床開発とは、製薬会社や医療機器メーカー、CRO(開発業務受託機関)などが医薬品の承認のために必要なデータをそろえる活動のことです。具体的には市場調査・開発プランの作成から始まり、試験の立ち上げ、治験薬の有効性や安全性のモニタリング、得られたデータの評価・考察、規制当局への承認申請までの様々な活動が含まれます。
また、CRA(臨床開発モニター)は別名「治験モニター」とも呼ばれます。治験とは、新しい医薬品の開発に必要なデータを収集するための試験のことです。具体的には、製薬メーカーが開発した新しい薬(治験薬)を患者や健康な人に投与し、その効果や安全性を検証するための試験のことです。この試験を通じて、厚生労働省から医薬品としての承認を得ることが目的です。
面接の際には、臨床開発や治験について説明を求められることも多いです。このページでしっかり勉強しておきましょう。
 臨床開発とは?
臨床開発とは?
臨床開発とは、製薬会社や医療機器メーカー、CRO(開発業務受託機関)などが新薬を販売するために必要なデータをそろえる活動のこと。
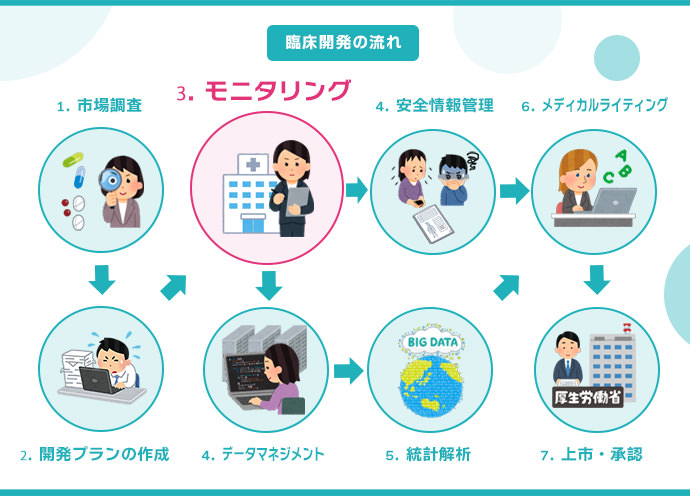
製薬会社や医療機器メーカー、CRO(開発業務受託機関)などが新薬を販売するために必要なデータをそろえる活動を臨床開発と言います。臨床開発は、市場調査や開発計画の作成から始まり、試験の立ち上げ、モニタリング、治験薬や医療機器の有効性および安全性の評価、規制当局への承認申請までのさまざまな活動が含まれます。
臨床開発に携わる人としては、医療機関を訪問して治験のデータを収集するCRA(臨床開発モニター)が知られていますが、市場調査をサポートする薬事コンサルタント、治験実施計画書の作成を担当する企画開発担当者、治験で得られたデータの管理や分析を行うDM(データマネージャー)や統計解析担当者、副作用や安全性に関する情報を収集するPV(安全性情報担当者)、承認申請に必要な書類などを作成するMW(メディカルライター)など、さまざまな専門家が協力して行っています。
以下では、それぞれの項目について解説します。
臨床開発のプロセスと担当者 新薬の開発プロセス 治験とは 治験のプロセス 治験の位置づけ 治験の成功率 新薬の上市数の推移 治験が行われている領域 治験が抱える問題点 治験の規則 治験と治療の違い 治験参加のメリット インフォームド・コンセント
臨床開発のプロセスと担当者
臨床開発は、市場調査 → 開発プランの作成 → モニタリング → データマネジメント → 生物統計解析 → 安全情報管理 → メディカルライティング → 承認/上市 の順に進められる。
- 市場調査
- 新薬の市場性を調査します。
 患者や医療従事者のニーズ、競合製品の動向などを調査します。その後、市場調査で得られたデータを分析し、法律や規制に沿った新薬の開発戦略を立案します。
患者や医療従事者のニーズ、競合製品の動向などを調査します。その後、市場調査で得られたデータを分析し、法律や規制に沿った新薬の開発戦略を立案します。- 市場調査をサポートする人を薬事コンサルタントと言います。薬事コンサルタントには、医薬品や医療機器の開発に関する法律や規制の知識、PMDAとの折衝能力、問題解決能力、プロジェクトマネジメント能力、英語力などの要素が必要とされています。
- 開発プラン
の作成 - 治験実施計画書などを作成します。
 治験を実施するための計画書を作成します。計画書には、治験の目的、方法、期間、予算など、病院や製薬会社が遵守しなければならない治験に関する全ての要件が記載されています。
治験を実施するための計画書を作成します。計画書には、治験の目的、方法、期間、予算など、病院や製薬会社が遵守しなければならない治験に関する全ての要件が記載されています。- 治験実施計画書は、企画開発担当者が中心となって作成します。企画開発担当者には、医薬品や医療機器の開発に関する最新の情報の把握、市場動向の分析力、戦略や計画を立案する能力、プレゼンテーション力、調整力、交渉力、創造力、英語力などの要素が必要とされています。
詳しくはこちら治験実施計画書の作成
- モニタリング
- 治験の進行状況を確認します。
 治験が治験実施計画書などを遵守して適切に実施されているかを確認します。
治験が治験実施計画書などを遵守して適切に実施されているかを確認します。- 治験のモニタリングを行う人をCRA(臨床開発モニター)と言います。CRA(臨床開発モニター)には、治験の実施に関する知識、コミュニケーション能力、正確性、調整力、英語力などの要素が必要とされています。
詳しくはこちらCRAとは
- データ
マネジメント - 治験で収集されたデータを管理します。
 治験で収集されたデータの入力や検証、クエリー(照会)処理、データベース設計・バリデーションなどを行い、データの正確性と整合性を確保します。
治験で収集されたデータの入力や検証、クエリー(照会)処理、データベース設計・バリデーションなどを行い、データの正確性と整合性を確保します。- データマネジメントを行う人をDM(データマネジメント担当/データマネージャー)と言います。DM(データマネジメント担当/データマネージャー)には、データを収集・管理・検証する能力、統計的手法に関する理解、注意力、ITスキルなどの要素が必要とされています。
詳しくはこちらDMの仕事内容
- 生物統計解析
- 臨床試験で収集されたデータを統計的に解析します。
 治験で収集されたデータを統計的に解析し、被験薬の有効性や安全性に関する仮説を検証したり、被験薬と既存薬との比較を行い有効性や安全性などを評価します。
治験で収集されたデータを統計的に解析し、被験薬の有効性や安全性に関する仮説を検証したり、被験薬と既存薬との比較を行い有効性や安全性などを評価します。- 生物統計解析を行う人を統計解析担当者と言います。統計解析担当者には、統計学や数学の知識、プログラミングスキル、論理的思考力、ITスキルなどの要素が必要とされています。
- 安全情報管理
- 治験中に発生した副作用や安全性に関する情報を収集・管理します。
 治験中に発生した副作用や安全性に関する情報を収集・管理し、有害事象や副作用の頻度や重症度を判断したり、治験実施計画書や被験者同意書などの文書の更新、副作用報告書の作成、治験を中止するかの判断などを行います。
治験中に発生した副作用や安全性に関する情報を収集・管理し、有害事象や副作用の頻度や重症度を判断したり、治験実施計画書や被験者同意書などの文書の更新、副作用報告書の作成、治験を中止するかの判断などを行います。- 治験の安全情報管理を行う人をPV(安全性情報担当者)と言います。PV(安全性情報担当者)には、副作用評価を適切に行うための医学薬学知識、安全性評価の知識、情報を収集・評価・報告する能力、英語力、スケジュール管理能力などの要素が必要とされています。
詳しくはこちらPVの仕事内容
- メディカル
ライティング - 治験の結果を報告書にまとめます。
 治験結果に基づき、報告書や承認申請資料、学術論文やポスターなどを作成します。
治験結果に基づき、報告書や承認申請資料、学術論文やポスターなどを作成します。- 承認申請に必要な文書を作成する人をメディカルライター(MW)と言います。メディカルライター(MW)には、医薬品や医療機器の開発に関する文書を作成する能力、医学や薬学の知識、文章力、英語力などの要素が必要とされています。
- 承認/上市
- 新薬の販売に必要な承認手続きを行います。
 新薬の販売に必要な承認手続きを厚生労働省に申請します。
新薬の販売に必要な承認手続きを厚生労働省に申請します。- 厚生労働省は、被験薬の有効性や安全性に関するデータや文書を審査し、承認基準に適合しているかを判断します。審査で承認されれば、新薬として販売(上市)が可能となります。
- 承認申請を行う人を薬事担当者と言います。薬事担当者には、薬事法や関連法規の知識、文書作成能力、コミュニケーション力、英語力などの要素が必要とされています。
 新薬の開発プロセス
新薬の開発プロセス
新薬の開発には10年以上の長い期間と1,000〜2,000億円の巨額な費用がかかる。
日本では毎年、約30~50種類の新薬(NMEのみ、全承認品目数はこの数のおよそ3倍ほど)が誕生しています。新薬の開発には10年以上の長い期間と1,000〜2,000億円の巨額な費用がかかると言われています。最初にスクリーニングされた物質が実際に薬となる確率は約2万~3万の1と言われています。
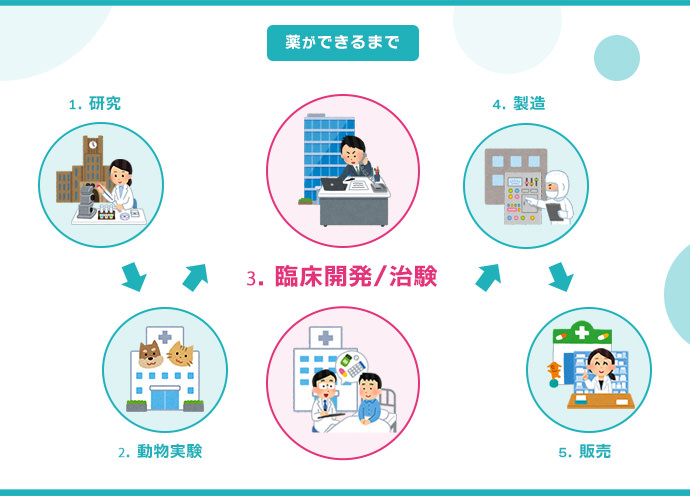
新薬は以下のようなプロセスを経て完成します。
- 基礎研究
(2~3年) - 自然界から薬になる可能性のある物質を探したり、科学的に作り出したりします。
- 最近はバイオテクノロジーやAIの進歩により、コンピューターを使用して物質の立体構造を予測し、薬の候補を作ることができるようになってきました。
 その物質が薬になるかどうかを、試験管の中で実験します。
その物質が薬になるかどうかを、試験管の中で実験します。
- 前臨床試験
(3~5年) - 動物で試験を行います。
 ネズミやウサギ、イヌ、サルなどの動物を使って、薬の候補が動物の体内でどのように吸収、分布、代謝、排泄されるかを調べます。そして、薬として期待される効果があるかどうか、副作用はないかなどを検討し、候補を絞り込んでいきます。
ネズミやウサギ、イヌ、サルなどの動物を使って、薬の候補が動物の体内でどのように吸収、分布、代謝、排泄されるかを調べます。そして、薬として期待される効果があるかどうか、副作用はないかなどを検討し、候補を絞り込んでいきます。- 安全性試験の実施方法や記録については、その試験結果の信頼性を確保するためにGLP(Good Laboratory Practice)という実施基準が定められています。日本では、「医薬品の安全性に関する非臨床試験の実施基準に関する省令」が厚生労働省から通達されています。
- 治験
(3~6年) - 人間で試験を行います。
 動物で試験した結果、効果と安全性が確認された物質だけが新しい薬の候補となり、人間での臨床試験に進むことができます。これが「治験」です。詳しくは以下で説明しますが、CRA(臨床開発モニター)が関わる部分になります。
動物で試験した結果、効果と安全性が確認された物質だけが新しい薬の候補となり、人間での臨床試験に進むことができます。これが「治験」です。詳しくは以下で説明しますが、CRA(臨床開発モニター)が関わる部分になります。
詳しくはこちら治験の流れ

- 製造販売後
臨床試験
(2~10年) - 薬が市販された後、より多くの患者の治療に使用された際の効果や安全性を確認します。
 治験と呼ばれませんが、治験と似た規則のもとで行われます。
治験と呼ばれませんが、治験と似た規則のもとで行われます。
 治験とは
治験とは

治験とは、新しい薬を開発する際に、厚生労働省から薬としての承認を受けるために必要なデータを収集する臨床試験のこと。
新しい薬を開発するためには、動物で薬の候補となる物質の効果や毒性を調べるだけでなく、人間での有効性(効果)や安全性(副作用)を確認する必要があります。
人間での有効性や安全性について調べる試験を「臨床試験」と呼びますが、その中でも医薬品の承認のために必要なデータを収集する臨床試験のことを「治験」と呼びます。治験は厚生労働省が定めた規則(医薬品の臨床試験の実施基準:GCP)に従って行われます。
治験を行う医師は、患者に治験の内容を詳しく説明し、患者は治験の内容を十分に理解した上で、自らの意思で治験への参加に同意することが必要です。
 治験の定義
治験の定義
治験とは、薬機法(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律)の第2条で、「治験とは、承認申請に提出すべき資料のうち、臨床試験の試験成績に関する資料の収集を目的とする試験の実施をいう」と定義されています。
また、第80条の2において、「厚生労働省で定めた基準に従って、治験を依頼・実施・管理しなければならない」と治験の取扱いについて定められています。
治験のプロセス
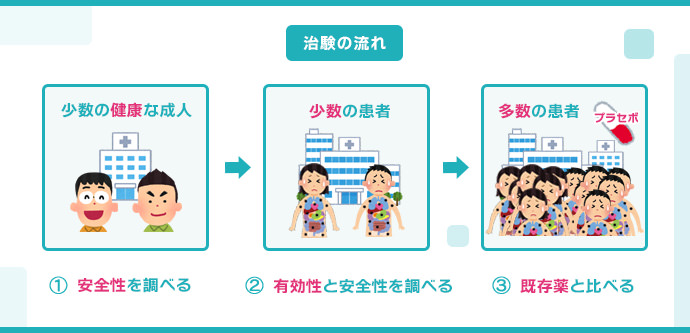
治験は以下の3つの段階に分かれており、各段階で薬の安全性や有効性を検証します。各段階(第一相、第二相、第三相)の被験者数の比率はおおよそ1:3:6で、後の段階に進むほど人数が増えます。
- 第一相試験
- 主に健康な男性を対象とした試験です。20~100人程度の健康な成人を対象に、ごく少量から徐々に薬の候補物質の投与量を増やし、主に安全性を調査します。
- 動物での実験結果があっても、人間を対象に再度調べることが必要です。動物では効果が見られたとしても、、人間でその効果が見られない場合や、逆に、動物では見られなかった副作用が人間で現れることがあります。
- 人体における安全性が未確認であるため、通常は妊娠の可能性のある女性は参加できず、主に男性が対象とされます。米国ではFDAの症例により女性の被験者が増加していますが、日本では女性の被験者の割合は10〜20%台にとどまっています。
- がんなどの治療不能な重篤な疾患の第一相試験では、被験者は初めから患者となります。
- 治験専門クリニックで行われることが多く、平日だけでなく、多くの被験者が休日を取得できる土日にも実施されます。
- 第二相試験
- 通常、200~300人程度の少数の患者を対象とした試験です。薬の候補物質が効果を示すと予測される比較的少人数の患者を対象に、有効性、安全性、用法・用量を調査します。
- 第三相試験の次にCRA(臨床開発モニター)が担当することが多い試験です。大学病院などの大規模な病院で行われることが多いです。
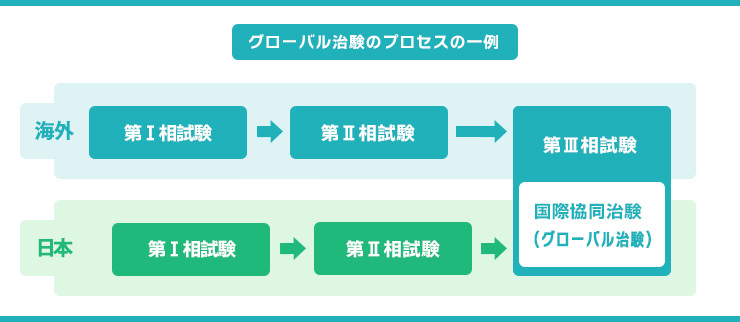
- 国際共同治験の場合、海外のデータを利用して一部を省略できる場合があります。
- 第三相試験
- 大勢の患者を対象とした試験です。実際の治療に近い状況で薬の有効性、安全性、用法・用量を確認し、既存薬との比較を行います。
- 数百~数千人の患者を対象とすることが多いです。大規模グローバル試験では、数万人から数十万人の患者を対象とする場合もあります。
- プラセボ(偽薬)を使用し、データの偏りを最小限に抑える「二重盲検ランダム化比較試験」が基本的に行われます。
- CRA(臨床開発モニター)が主に担当するのはこの第三相試験です。 大学病院から専門クリニックまで、多くの全国の医療機関で治験が行われます。
- 第四相試験
(製造販売後
臨床試験) - 厚生労働省の承認を受けた後に行われる、薬の安全性を確認するための試験です。GPSP省令・GVP省令に基づき実施され、市販後臨床試験とも呼ばれます。市販後調査(PMS)を通じて行われることが多いです。
- この段階は、治験とは呼ばれません。
- 市販後調査(PMS)は、新薬投与後の患者の状態を6ヶ月間に渡って調査する「市販直後調査」、治験の対象者とならなかった患者の有効性・安全性を調査する「特定使用成績調査」、長期的な使用実態を調査する「使用成績調査」に分類されます。製薬会社のMRやモニターが医師を訪問し、情報収集を行うことが多いです。
 治験の位置づけ
治験の位置づけ
人を対象とする医学的研究を「臨床研究」と言い、臨床研究のうち薬や治療法の効果や安全性を調べる試験を「臨床試験」と言う。臨床試験のうち医薬品や医療機器などを厚生労働省に承認してもらうために行う試験を「治験」と言う。
CRA(臨床開発モニター)は主に治験に関わりますが、特定臨床研究にも携わることがあります。
携わる業務範囲を明確に区別するため、治験に携わっている人を「臨床開発モニター」「治験モニター」、臨床研究に携わっている人を「臨床研究モニター」、製造販売後臨床試験や市販後調査に携わっている人を「PMSモニター」と呼び分ける場合があります。逆に業務範囲を区別しないときは「モニター」とだけ呼ぶこともあります。
- モニターのキャリアパスはこちら

| 種類 | 内容 | 職種 |
| 臨床研究 | 患者の病気の原因解明や治療の改善を目的とした研究。患者が直接研究に参加する「介入研究」と、検査データや血液サンプルの提供などを依頼する「観察研究」に分けられる。市販後調査(PMS)も含まれる。 | 臨床研究モニター、PMSモニター、MR |
| 臨床試験 | 臨床研究のうち、薬や治療法などの安全性と有効性を評価することを目的とした研究。製造販売後臨床試験(市販後臨床試験)も含まれる。関連法規はGCP省令、GPSP省令。 | 臨床研究モニター、PMSモニター |
| 特定臨床研究 | 臨床研究のうち、製薬企業から資金提供を受けるもの、または未承認薬(日本で承認されていない薬)あるいは適応外薬(ある疾患に対しては承認されているが別の疾患への効能は承認されていない薬)を使用するもの。関連法規は臨床研究法。 | 臨床研究モニター |
| 治験 | 臨床試験のうち、薬や医療機器などを厚生労働省に承認してもらうための試験。この試験は、企業が主導する規模の大きな「企業治験」と、医師が主導する規模の小さな「医師主導治験」に分けられる。関連法規は薬機法、GCP省令。 | 臨床開発モニター(治験モニター) |
- 臨床開発モニターと臨床研究モニター(またはPMSモニター)の違いはこちら
治験の成功率
治験の成功率は約6~7%。特にがん(オンコロジー)領域の治験の成功率は3~4%と低い。
治験が成功する確率は、約6~7%と低いです。これは、残りの93~94%が失敗に終わることを意味します。
治験の各段階における成功率は低く、第1相は約47%、第2相は約28%、第3相は約55%であり、承認申請後に市販される確率は約92%です。
治験の成功率
※成功確率は前臨床試験を終えた化合物を100とした場合
※成功確率はNorstellaなどの論文を参考に作成。
新薬の上市数の推移
2021年に世界で販売が開始された新薬(NAS)の数は、5年前と比べて2倍(過去最多)を記録した。
2021年に世界で販売が開始された新薬(NAS)の数は、パンデミックの影響もあり5年前と比べて2倍に増加し過去最多を記録しました。しかし、日本で販売が開始された新薬(NME)の数は2014年に過去最多を記録して以来、更新されていません。そのため、2017年以降、日本で販売される新薬の数は、世界との差が広がったままです。
治験が行われている領域
治験が最も多く行われている領域はがん(オンコロジー)。コロナの影響で一時的にワクチンの治験も増加。
治験が最も多く行われている領域はがん(オンコロジー)です。従来の抗がん剤のほかに、分子標的薬や免疫チェックポイント阻害薬など、さまざまな新薬が開発されています。その結果、がん(オンコロジー)領域の治験数は大きく増加しました。
薬物の治験計画届出件数の推移
※PMDAより出典
治験が抱える問題点
新薬の審査期間の短縮や国際共同治験の増加により、多くのドラッグ・ラグが解消され、2010年代前半までには治験の空洞化が大幅に改善された。しかし・・・
2010年より前は、日本の新薬の審査期間(審査ラグ)が主要国の中で最も長かったのですが、現在はトップクラスの短さとなっています。
実際に、CIRSの調査によれば、2009年時点で日本の新薬の審査期間は650日前後と長く、主要国の中では最下位でした。しかし、2010~2012年ごろにPMDA(医薬品医療機器総合機構)の審査官を増員するなどの取り組みの結果、2014~2016年や2022年には主要国の中で最も短かくなりました。
志望動機に「治験コーディネーター(CRC)になってドラッグ・ラグ(審査ラグ)を解消したい」と書く人もいますが、ドラッグ・ラグ(審査ラグ)の問題はすでに大幅に改善されているという事実を踏まえて、志望動機を見直すことをおすすめします。
日本の治験開始が海外に比べて遅れる開発ラグについても、2018年には国際共同治験の割合が50%を超え、製薬メーカーの合併も相次ぎ、グローバル化が進んだ結果、治験の空洞化はほぼ解消さると思われましたが・・・
審査期間の推移
※CIRSを参照。
国際共同治験の推移
※独立行政法人医薬品医療機器総合機構 事業年度業務報告書をもとに作成。2022年の件数が跳ね上がっているのは様式改正のため。
患者数の少ない希少がんやゲノム医療、バイオテクノロジーといった先端分野で、新たなドラッグ・ラグと言われるドラッグ・ロス問題が発生!
2010年代後半からは、解消されたと思われたドラッグ・ラグが再び問題化しています。その原因は、薬価制度の見直しを繰り返した結果、日本の新薬開発市場への魅力が失われ、患者数の少ない希少がんやゲノム医療、バイオテクノロジーといった先端分野で、新薬が日本で開発されないケースが増えているからです。
特に抗がん剤という開発数の最も多い分野では、新興企業からの製品の増加により、日本での治験を省略するドラッグ・ロスが大幅に増加しています。
実際に、2016~2020年に米FDA(米国食品医薬品局)が承認した60品目の抗がん剤のうち、41品目が日本で未承認となっており、これは全体の約68%を占めます。また、未承認となった41品目のうち、約半数の22品目はバイオベンチャーなどの新興企業が開発した製品です。出典:ドラッグ・ラグ:なぜ、未承認薬が増えているのか?)
2023年末のデータでも米国FDA承認品目243に対して、日本未承認品目は164となっており、未承認割合は67.5%と高いままです。疾患別では抗悪性腫瘍剤が49品(74.2%未承認)で最多のままです。出典:国内未承認薬の最新動向)
このように、日本では先端分野の新薬開発に対するインセンティブが低く、海外で承認されている新薬が日本で承認されないまま放置されることで、患者にとって大きな不利益となっています。
ドラッグ・ロスを解消するためには、日本の製薬メーカーや研究機関が先端分野の新薬開発に積極的に取り組むことが必要です。また、政府や行政も薬価制度や規制環境を見直し、新薬開発市場へのインセンティブを高めることが求められます。
ドラッグロス解決に向けた最新動向
- 2024年度薬価制度改革で「革新的新薬迅速導入加算」がスタート。海外ベンチャーも対象。
- PMDAが国際共同治験ワーキングシェア型審査(ICMRA Pilot)に参画、参加費を半額に。
- 厚労省は未承認薬86品の優先リストを作成し、AMED支援でアカデミア治験を補助。など
- 2025年の薬機法改正で革新的医薬品実用化支援基金の設立について法律に明記
- 2025年の薬機法改正で条件付き承認制度の対象を「臨床的有効性が合理的に予測可能な場合等」に拡大
国別の未承認薬の割合
※2010~2021年の481品目を対象 ドラッグ・ラグ:日本と欧州の未承認薬状況の比較より出典。
企業別の国内承認率の比較
※2010~2021年 ドラッグ・ラグ:日本と欧州の未承認薬状況の比較より出典。
 治験の規則
治験の規則
治験は厳格な規則(GCP)に従って実施される。
治験を実施する際には、参加者の人権と安全性が最大限に保護されることが求められます。このため、「薬機法(旧薬事法)」という法律および厚生労働省が定めた厳格な基準(医薬品の臨床試験の実施基準:GCP)に基づき、治験の進行に関する厳格な規則が策定されています。主要な規則は以下の通りです。
治験を実施する際の規則
- 治験の内容を国に届け出る
- 治験審査委員会で治験の内容を事前に審査する
- 治験に参加することに同意した患者だけを治験に参加させる
- 重大な副作用を国に報告する
- 製薬会社は治験が適切に進行しているかを確認する
別の記事でも詳しく説明していますので、さらに知りたい方はそちらもご覧ください。
詳しくはこちらGCPについて
 治験と治療の違い
治験と治療の違い
治験と治療は異なる点がある。
治験と一般的な治療とはどのような違いがあるのでしょうか。以下にいくつかの主な点をまとめましたので、ご確認ください。
| 治験 | 治療 | |
| 厚生労働省の承認を得ていません。※プラセボを除く | 使用される薬 | 厚生労働省の承認を得ています。 |
| 試験が主な目的です。ただし、治療的な側面もあります。 | 薬を飲む目的 | 治療が主な目的です。 |
| 患者が希望しても、指定された病院でしか受けられません。 | 病院 | 患者が希望する病院であれば、どこでも治療を受けることが可能です。 |
| 治験担当医師(複数の場合もあります)が一貫して診察します。治験担当医師以外が診察することはありません。 | 医師 | 主治医以外の医師が診察することもあります。 |
| 一般的な治療よりも長い時間がかかります。 | 診察時間 | 一般的には短時間で終わります。 |
| 一般的な治療よりも細かな検査が行われます。 | 検査内容 | 健康保険の適用範囲内で行われる検査が主となります。 |
 治験参加のメリット
治験参加のメリット

治験に参加すると、治験内容や医療機関の体制によっては多少の違いがありますが、おおむね以下のようなメリットが得られます。
治験に参加するメリット
- これまでにない新しい治療を受けるチャンスがあります。
- 経験豊富な治験担当医師による丁寧な診察を受けることができます。
- 一般の診療に比べて、より細かな検査が行われるため、ご自身の病気の状態を詳しく知ることができます。
- 治験薬の費用や、治験中の検査費用を支払う必要がありません。
そして、何よりもかけがえのないことは、「次の世代により良い薬を残すために協力する」という社会貢献ができることです。
 インフォームド・コンセント
インフォームド・コンセント

インフォームド・コンセントとは、治療を受ける前に自分の病気やその治療方針について医師やその他の関係者から十分な説明を受け、患者が説明の内容を十分に理解し納得した上で、患者自身の意思で治療に同意することです。
インフォームド・コンセントは一般的な治療でも行われますが、治験は試験的な性質があるため、参加者の人権を尊重し安全性を確保するためにも欠かせない手続きです。
インフォームド・コンセントで説明される主な内容は以下の通りです。
インフォームド・コンセントで説明される主な内容
- 治験の目的、治験薬の使用方法、検査内容、参加期間
- 期待される効果と予想される副作用
- 治験への参加はいつでもやめることができ、不参加でも不利益は受けないこと
- 副作用が起きて被害を受けた場合、補償を求めることができること
- 担当する医師の氏名や連絡先
- 治験に関する質問や相談のための問い合わせ先
別の記事でも詳しく説明していますので、さらに知りたい方はそちらもご覧ください。
詳しくはこちらインフォームド・コンセントとは











転職サポート申し込みはこちら
- ステップ1
- まずは申し込み。入力は1分で終わります。
- ステップ2
- 希望にマッチした求人情報を提供します。
- ステップ3
- 書類選考・面接
- ステップ4
- 内定・入社
- 入社後もずっとサポート!

 年収査定はこちら
年収査定はこちら
- 転職を考える際、最も重要な条件の一つは給与です。
CRA(臨床開発モニター)への転職を考えている方々にとって、自身のキャリアや経験がどの程度評価されるのか、気になることでしょう。
こちらでは、あなたのプロフィールに基づき、CRA(臨床開発モニター)へ転職した場合の年収を予測します。

 合格予想はこちら
合格予想はこちら
- 「社会人経験が少ない」「転職回数が多い」といった理由で、選考に通過できるか不安になり、応募をためらう方も多いと思います。
こちらでは、あなたのプロフィールに基づき、CRA(臨床開発モニター)に応募した場合の書類選考の通過率や面接の合格率を予測します。

 掲示板で質問をする
掲示板で質問をする
- 些細な悩みや、ふとした疑問がある場合は、掲示板で気軽に質問しましょう。
面倒な登録は必要ありません。匿名で簡単に質問できます。多くの人の協力を得て、あなたの疑問を解決しましょう。
CRA(臨床開発モニター)や人事担当者などの専門家が回答いたします。








 CRA
CRA

 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAに
CRAに
 CRAの
CRAの
 CRO
CRO
 CRO
CRO
 臨床開発
臨床開発
 製薬会社と
製薬会社と
 CROから
CROから



 2027年4月からの転職
2027年4月からの転職 CRA未経験特集
CRA未経験特集 薬剤師特集
薬剤師特集 MR特集
MR特集 看護師特集
看護師特集 臨床検査技師特集
臨床検査技師特集 保健師特集
保健師特集 獣医師特集
獣医師特集 理系大卒・院卒特集
理系大卒・院卒特集 CRC経験者特集
CRC経験者特集


 求人検索
求人検索  ログイン
ログイン 会員さま専用
会員さま専用 CRAの仕事
CRAの仕事  臨床開発業界の研究
臨床開発業界の研究 経験・資格別の注意点
経験・資格別の注意点 応募書類の作成
応募書類の作成 面接・適性検査の対策
面接・適性検査の対策 みんなのクチコミ
みんなのクチコミ みんなの質問と回答
みんなの質問と回答 転職成功事例
転職成功事例 マンガで分かるCRA
マンガで分かるCRA 便利な機能
便利な機能 相談/年収査定/合格予想
相談/年収査定/合格予想 2027年から働くには?
2027年から働くには? 退職手続き
退職手続き 開催中のキャンペーン
開催中のキャンペーン 《CRAばんく》とは
《CRAばんく》とは