


PV・安全性情報担当者の仕事内容
 PV(安全性情報担当者)とは
PV(安全性情報担当者)とは
PV(安全性情報担当)とは、患者の安全を守るために、薬の副作用などの情報を集めて分類する専門職のことです。
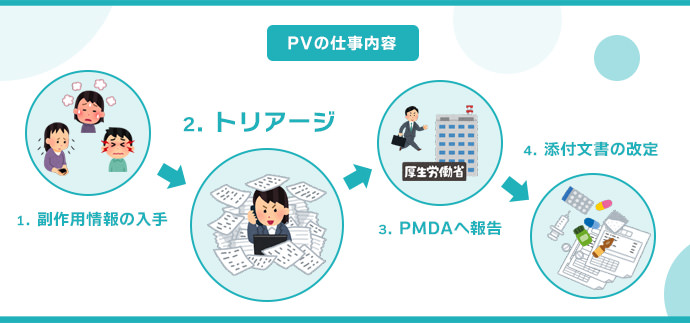
PV(安全性情報担当者)の仕事内容を分かりやすく説明すると、「副作用を重要度に応じて分類し、所定のフォームに入力する業務」となります。
PV(安全性情報担当者)には、医学や薬学に関する幅広い専門知識に加え、英文の読解力が求められます。また、定型業務の割合が高く、ルーティンワークの要素が強い職種です。
以下では、PV(安全性情報担当者)の仕事内容について詳しく解説します。
PV(安全性情報担当者)の仕事内容
 リスク管理計画の策定
リスク管理計画の策定
市販後の医薬品のリスクを低減するための医薬品リスク管理計画(RMP)※1 を、PMDAと協議して決定します。治験の場合はプロトコール(治験実施計画書)に、安全性情報の収集と評価の方法を記載します。
※1 RMP(Risk Management Plan)・・・医薬品リスク管理計画のこと。「安全性検討事項(Safety Specification)」「医薬品安全性監視活動(Pharmacovigilance Plan)」「リスク最小化計画(Risk Minimisation Plan)」の3要素で構成される。

医薬品と治験薬で、PVの仕事の流れは異なります。
医薬品の方が使用者が多いため、有害事象の報告件数も多くなります。そのため、PVは一般的に、市販後医薬品の安全性情報担当者を指すことが多いです。
医薬品の方が使用者が多いため、有害事象の報告件数も多くなります。そのため、PVは一般的に、市販後医薬品の安全性情報担当者を指すことが多いです。
 海外医薬品安全性情報報告書の翻訳
海外医薬品安全性情報報告書の翻訳

MedWatch※2 やFDA Adverse Event Reporting System(FAERS)※3 などの海外医薬品安全性情報報告書を、日本語に翻訳します。
翻訳には、英語力だけでなく、医学や薬学に関する幅広い専門知識が必要となります。機械翻訳ツールを活用することも多いですが、専門家によるレビューは欠かせません。
※2 MedWatch・・・米国食品医薬品局(FDA)の安全性情報および有害事象報告プログラムのこと。医療における有害事象に関するデータを収集している。
※3 FDA Adverse Event Reporting System(FAERS)・・・米国食品医薬品局(FDA)に報告された医薬品の副作用情報を集積したデータベースのこと。

海外の安全性情報の文書の翻訳は、PVではなく、医学・薬学の専門知識と高度な英語力や翻訳スキルのあるメディカルライターが担当することが多いです。
 用語の調査
用語の調査

MedDRA※4 などのデータベースを活用し、用語の調査を行います。MedDRAを使用することで、異なる国や地域で収集された情報を比較分析することができます。
海外の報告書には、日本とは異なる用語や基準が使用されている場合があるため注意が必要です。必要に応じて、海外の規制当局や医療機関に問い合わせて、情報の確認を行います。
※4 MedDRA・・・医薬品安全性情報に関する約9万種類の用語が収録されている国際的な用語標準のこと。
 有害事象報告書や医薬品安全性情報報告書の受領
有害事象報告書や医薬品安全性情報報告書の受領

患者や医療機関から有害事象報告書や医薬品安全性情報報告書などを受け取り、内容に不備がないか確認します。不備がある場合は、報告者に追加情報を求めます。
なお、治験を実施する際に報告する有害事象の書類は「有害事象報告書」、既に市場に出ている医薬品の副作用を報告する書類は「医薬品安全性情報報告書」と呼ばれます。
 2020年に治験副作用報告の範囲が拡大された
2020年に治験副作用報告の範囲が拡大された
2020年9月1日に施行された改正薬機法により、治験における有害事象の報告義務が拡大され、被験薬だけでなく対照薬や併用薬による有害事象についても、治験依頼者が厚生労働大臣(PMDA)へ報告する義務が明確化されました。治験のPV(安全性情報担当者)の業務量が増加した結果、求人も増加しました。
 副作用や有害事象のトリアージ(選別・評価)
副作用や有害事象のトリアージ(選別・評価)

厚生労働省の副作用の重篤度分類基準、CTCAE※5 やJADER※6 、などに基づき、副作用や有害事象を、重症度・因果関係・発生頻度などを考慮して多段階に選別・評価(トリアージ)します。
トリアージの結果に基づいて、報告書を
1)重篤な副作用や、新たな副作用、集団発生などが疑われるPMDAへの報告が必要な事象
2)軽度~中程度の副作用であっても、注意喚起が必要と判断される社内共有が必要な事象
3)軽微な事象で報告や共有の必要がない事象
へと振り分けます。
※5 CTCAE・・・米国立がん研究所(NCI)が定める、臨床試験における有害事象の評価基準。重症度、因果関係、発生頻度などを5段階で評価する。
※6 JADER・・・PMDAが公開・運用する医薬品副作用報告データベース。国内の個別症例安全性報告(ICSR)を収載しており、副作用情報の検索・統計解析に用いられる。

PVの主な業務は、副作用や有害事象の情報を適切に分類して、データベースに入力することです。
 PMDAへの報告
PMDAへの報告

PMDA※7 への報告が必要な有害事象については、CIOMSフォーム※8や国内様式に従い、社内の安全性情報管理システムに患者情報、医薬品情報、有害事象情報などの必要な情報を入力します。その後、PMDAの副作用等報告受付ゲートウェイ経由で電子データを提出します。
PMDAへの報告期限は、事象の重症度によって異なります。特に、重篤な事象の場合は、速やかに報告する必要があります。これは、患者の安全を確保し、他の患者に同様の事象が発生するのを防ぐためです。
※7 PMDA(独立行政法人医薬品医療機器総合機構)・・・医薬品や医療機器などの承認審査や安全対策業務を行う、厚生労働省が管轄する独立行政法人。
※8 CIOMS(シオムス)・・・国際医学団体協議会の略称です。世界保健機構(WHO)と国際連合教育科学文化機関(UNESCO)によって1949年に設立された。CIOMSは有害事象報告書の書式の標準化を図り、一般に流布させた団体で、その書式は「CIOMS form」と呼ばれてる。

データの入力作業は派遣スタッフが行うことも多いです。その場合、PVは派遣スタッフの作業スケジュールを決め、的確な作業指示を行います。
 社内共有資料の作成
社内共有資料の作成
PMDAへの報告が必要ない事象であっても、事象の概要や原因、再発防止策などをまとめた資料を作成し、社内で共有します。
 安全性情報の更新
安全性情報の更新

収集された情報に基づいて、医薬品や治験薬の安全性情報を更新します。医薬品の添付文書の改訂が必要な場合は、PMDAに申請して承認してもらう必要があります。PV(安全性情報担当者)は添付文書改訂に伴うPMDAとの対応を行います。
治験薬の治験薬概要書などの修正が必要な場合も、PMDAに報告します。
また、安全性に関する緊急かつ重要な情報を、医療関係者や国民に迅速に伝えるために緊急安全性情報(イエローレター)や安全性速報(ブルーレター)の発行にも携わります。

医薬品の添付文書の改訂は、CROではなく製薬会社の安全性情報担当者が行うことが多いです。
 その他の業務
その他の業務
定期報告書の提出
安全性定期報告書※9、定期的ベネフィット・リスク評価報告書(PBRER)※10、年次報告書※11 などを、決められた時期に規制当局へ提出します。
※9 安全性定期報告書・・・新医療用医薬品について、リスク管理計画(RMP)の実施状況、製造販売後調査結果、副作用報告状況などを整理した日本独自様式の安全性報告。PBRERを添付すれば本様式の重複記載を省略できる。提出周期は承認(指定日)から最初の2年間は6か月ごと、以降は原則1年ごと。
※10 定期的ベネフィット・リスク評価報告書(PBRER)・・・医薬品の安全性や有効性に関するベネフィット・リスクの評価が記載された報告書。最初の2年間は6か月ごと、その後は原則1年ごとににPMDAへ提出。
※11 年次報告書・・・医薬品の安全性や有効性、製造販売状況、品質管理、広告宣伝などの情報が記載された報告書。毎年PMDAへ提出。
情報収集
薬物の安全性に関する最新の情報を収集します。情報収集には、国内外の学術論文、医薬品情報提供書、医薬品添付文書、規制当局のホームページなどを利用します。
文書作成
安全性情報報告書、社内共有資料、教育資料など、さまざまな文書を作成します。
コンプライアンス
医薬品に関する法規制を遵守し、コンプライアンスを徹底します。GCP(Good Clinical Practice)※12、GVP(Good Pharmacovigilance Practices)※13、GMP(Good Manufacturing Practice)※14 などのガイドラインを理解し、遵守する必要があります。
※12 GCP・・・治験の質を確保するための基準のこと。
※13 GVP・・・医薬品、医薬部外品、化粧品、医療機器の適正使用情報の収集や検討、市販後の安全確保措置の実施に関する基準のこと。
※14 GMP・・・医薬品製造工場における製造管理と品質管理の品質を確保するための基準のこと
 PV(安全性情報担当者)に必要なスキル
PV(安全性情報担当者)に必要なスキル
医学薬学に関する専門知識
医薬品の作用機序、副作用、臨床試験など、医学薬学に関する幅広い知識が必要です。特に、人の集団における薬物の使用とその効果や影響を研究する薬剤疫学は、市販後医薬品の適正使用の確立に役立ちます。
英語力
海外の安全性情報や学術論文に目を通したり、機会翻訳された内容のレビューを行うため、医療英語のReading力が必要です。
情報収集・分析能力
膨大な量の情報から、必要な情報を的確に収集し、分析する能力が必要です。
コミュニケーション能力
医療従事者、社内関係者、規制当局など、様々な関係者とコミュニケーションを図る能力が必要です。
文書作成能力
医学薬学に関する専門知識を活かした、わかりやすい文書を作成する能力が必要です。
倫理観
患者さんの安全を守るという倫理観に基づいて、業務を行う必要があります。
 PV(安全性情報担当者)のキャリアパス
PV(安全性情報担当者)のキャリアパス
製薬会社
・自社開発品の安全性に特化したキャリアを積むことができます。
・安全性部門だけでなく、数年おきに開発部門や製造部門などへのジョブローテーションが行われることが多いです。
CRO
・複数の製薬会社から委託される幅広い医薬品の安全性に関わることができます。
・将来的には、部門をとりまとめるリーダーやマネージャーなどのキャリアパスがあります。
規制当局
PMDAなどの規制当局で、医薬品安全性に関する審査・監督業務に携わることができます。
 PV(安全性情報担当者)のやりがい
PV(安全性情報担当者)のやりがい
患者さんの安全を守る仕事
PV(安全性情報担当者)は、医薬品の安全性情報を収集・分析・評価することで、患者さんの安全を守る重要な役割を担っています。
医薬品の開発に貢献する仕事
PV(安全性情報担当者)は、収集した安全性情報を基に、医薬品の改良や新たな医薬品の開発に貢献することができます。
グローバルな仕事
PV(安全性情報担当者)は、海外の製薬会社やCROと連携して、英語力を活かしてグローバルな業務に携わることができます。
 既存医薬品と治験薬のPV(安全性情報担当者)の仕事内容の違い
既存医薬品と治験薬のPV(安全性情報担当者)の仕事内容の違い
既存医薬品のPV(安全性情報担当者)
既存医薬品は安全性が広く認知され、豊富なデータが蓄積されています。規制当局へは定期的な報告が求められることが多いです。
そのため、既存医薬品のPV(安全性情報担当者)には、広範囲の情報収集力と大量のデータを効率よく処理できる能力が重要視される傾向があります。
治験薬のPV(安全性情報担当者)
治験薬は安全性は完全には確立されておらず、利用可能なデータも限られています。規制当局へは迅速な報告が求められることが多いです。
そのため、治験薬のPV(安全性情報担当者)には、最新の情報収集力と判断力、迅速にデータを処理できる能力が重要視される傾向があります。
よくある質問とみんなの回答
- Q
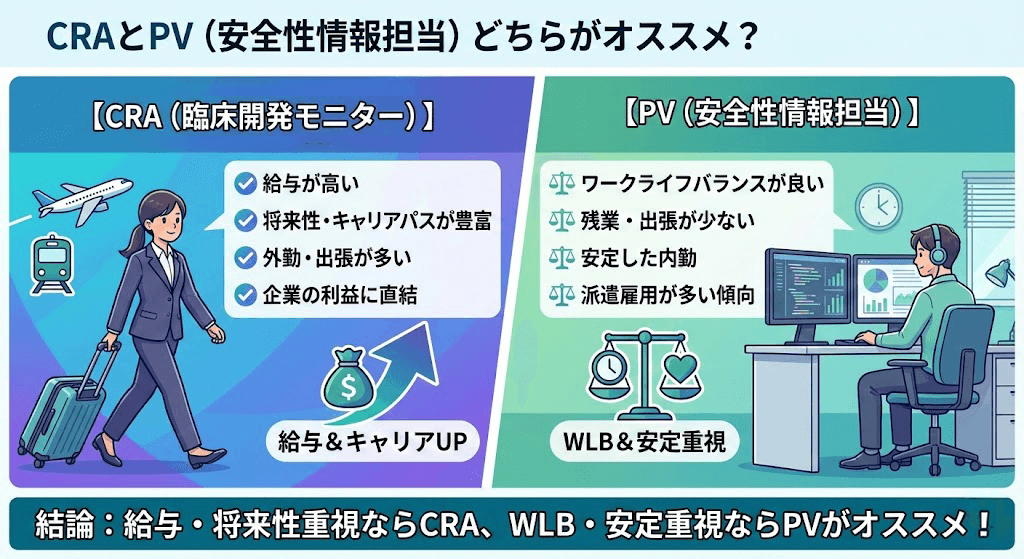
- CRAとPV(安全性情報担当)のどちらがオススメでしょうか。
- A
-
製薬会社で新薬開発に関わっています。...続きを見る
CRAとPVは仕事をする目的が異なります。CRAは新薬を患者様へ届けることが目的ですが、PVは新薬に限らず薬を安全に患者様に届けることが目的です。CRAは攻めでPVは守りです。仕事内容もCRAは華やかな一面があり、PVは地味で保守的です。
看護師としてのコミュニケーション力を活かせるのはCRAだと思います。CRAはCRCや医師、看護師などとの直接的なやりとりが発生します。PVは常に書類やデータとにらめっこですから、看護師としての経験は活かしづらいかもしれません。逆に看護師が向いていないと感じられているならPVのほうに適性があるかもしれません。
薬がなくて困っている患者様を助けたいなら新薬開発に携わることができるCRA、薬をできるかぎり安全に患者様に届けて副作用で困っている患者様を減らしたいならPVが良いのでは?
- Q
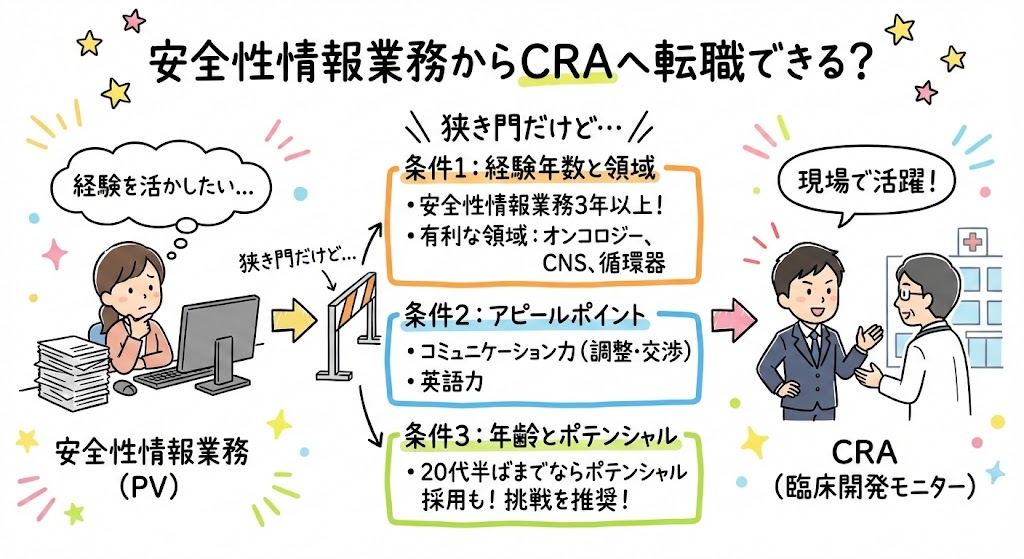
- 安全性情報業務に携わっていますがCRAへの転職は可能でしょうか。
- A
-
文系の大卒ですよね。微妙かもしれません。
安全性情報業務に携わられていた期間が3年以上あり、領域が多くの治験が行われているオンコロジーやCNS、循環器などでしたら可能性はありそうです。
あとはコミュニケーション力や英語力などをアピールしていくしかないと思います。
- Q
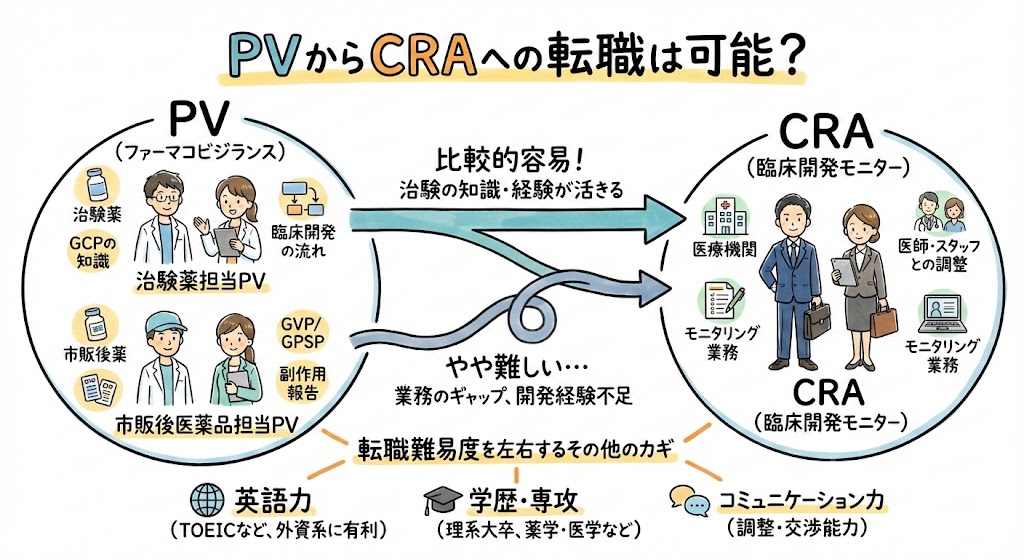
- PVからCRAへの転職は可能でしょうか
- A
-
PV(安全性情報担当者/安全性情報管理担当者)として担当している領域が、市販後医薬品か治験薬かによって、CRAへの転職難易度は異なります。...続きを見る
PVとして市販後医薬品を担当している場合、CRAへの転職はやや難しくなる可能性が高いです。
その理由は、必要とされる法規制の知識がGVPに関するものであり、主にやり取りする相手がMRや一般医師であること、そして扱う書類は医薬品安全性情報報告書や医薬品の添付文書など、医薬品開発や治験に直接関連しないものが多いからです。
一方で、PVとして治験薬を担当している場合、CRAへの転職はしやすくなる可能性が高いです。
その理由は、必要とされる法規制の知識がGCPに関するものであり、主にやり取りする相手は治験担当医師やCRA、CRCであること、そして扱う書類は有害事象報告書や治験実施計画書、治験薬概要書など、医薬品開発や治験に直接関連するものが多いからです。
質問者様の英語力や学歴、専攻分野、コミュニケーション力やCRAへの意欲の高さなどによっても、CRAへの転職のしやすさは異なりますが、PVとして担当している領域が、市販後医薬品か治験薬かによって、CRAへの転職のしやすさが変わることを知っておきましょう。
<役に立つ記事>
https://cra-bank.com/keijiban?gu=99
(CRAとPVのどちらがオススメでしょうか)
https://cra-bank.com/cratoha
(CRAとは)
https://cra-bank.com/cranoshigotonaiyou
(CRAの仕事内容とやりがい)
https://cra-bank.com/pvnoshigotonaiyou
(PV・安全性情報担当者の仕事内容)
https://cra-bank.com/keijiban?gu=39
(安全性情報業務に携わっていますがCRAへの転職は可能でしょうか)











 年収査定はこちら
年収査定はこちら
 合格予想はこちら
合格予想はこちら
 掲示板で質問をする
掲示板で質問をする







 CRA
CRA

 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAの
CRAの
 CRAに
CRAに
 CRAの
CRAの
 CRO
CRO
 CRO
CRO
 臨床開発
臨床開発
 製薬会社と
製薬会社と
 CROから
CROから



 2027年4月からの転職
2027年4月からの転職 CRA未経験特集
CRA未経験特集 薬剤師特集
薬剤師特集 MR特集
MR特集 看護師特集
看護師特集 臨床検査技師特集
臨床検査技師特集 保健師特集
保健師特集 獣医師特集
獣医師特集 理系大卒・院卒特集
理系大卒・院卒特集 CRC経験者特集
CRC経験者特集


 求人検索
求人検索  ログイン
ログイン 会員さま専用
会員さま専用 CRAの仕事
CRAの仕事  臨床開発業界の研究
臨床開発業界の研究 経験・資格別の注意点
経験・資格別の注意点 応募書類の作成
応募書類の作成 面接・適性検査の対策
面接・適性検査の対策 みんなのクチコミ
みんなのクチコミ みんなの質問と回答
みんなの質問と回答 転職成功事例
転職成功事例 マンガで分かるCRA
マンガで分かるCRA 便利な機能
便利な機能 相談/年収査定/合格予想
相談/年収査定/合格予想 2027年から働くには?
2027年から働くには? 退職手続き
退職手続き 開催中のキャンペーン
開催中のキャンペーン 《CRAばんく》とは
《CRAばんく》とは